La enfermedad de Wolman
La enfermedad de Wolman, descrita en 1954 por el médico Moshe Wolman, es una enfermedad pediátrica causada por la deficiencia de la enzima lisosomal lipasa ácida (AL). Esta enzima es una glicoproteína encargada de la hidrólisis de los ésteres de colesterol y triglicéridos en el paso de sangre periférica a la célula, particularmente cuando estos forman lipoproteínas de baja densidad (LDL). La deficiencia de esta enzima conduce a un almacenamiento masivo de triglicéridos y ésteres de colesterol. Estos depósitos dañan los tejidos afectados, principalmente el cerebro, hígado y bazo. Se incluye dentro del grupo de enfermedades ocasionadas por almacenamiento de lípidos, también llamadas lipidosis. La enfermedad de Wolman es uniformemente mortal a principios de la infancia, a menos que se realice tratamiento con trasplante de células hematopoyéticas (HCT). La enfermedad de Wolman es rara (menos de 80 casos identificados en PubMed).
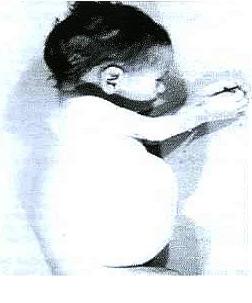
La mayoría de los pacientes cursan con un cuadro clínico similar que se caracteriza por vómitos, diarrea, esteatorrea, hepatoesplenomegalia, distensión abdominal y desnutrición progresiva. Los niños afectados son normales en el momento del nacimiento, pero pronto comienzan a presentar deterioro de las capacidades mentales, agrandamiento del hígado (hepatomegalia) y del bazo (esplenomegalia). También aparecen alteraciones intestinales como esteatorrea (eliminación de grasas por las heces) y distensión del abdomen. Otros síntomas comunes son anemia y depósitos de calcio en las glándulas suprarrenales. La muerte suele producirse antes del año de vida. Los cambios patológicos más significativos comprenden el depósito de triglicéridos y ésteres de colesterol en las células del sistema fagocítico, en el intestino delgado y la corteza adrenal.
No hay comentarios:
Publicar un comentario