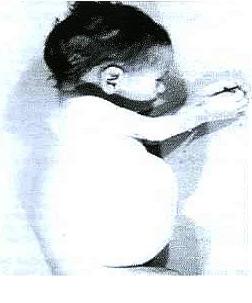
El síndrome de Bloom se caracteriza por hipersensibilidad a la luz solar, que da lugar a una erupción facial rojiza en la piel de las mejillas, además de en otras áreas expuestas al sol, como el dorso de las manos. Otros signos clínicos son:
- Telangiectasia;
- Baja estatura;
- Voz aguda;
- Distintos rasgos faciales, como caras estrechas y largas, micrognatismo de la mandíbula y nariz y orejas prominentes;
- Áreas hipo e hiperpigmentadas de la piel y manchas café con leche;
- Deficiencia de ciertas clases de inmunoglobulinas, lo que provoca neumonías recurrentes e infecciones de oído;
- Hipogonadismo con incapacidad para producir espermatozoides, por lo tanto, infertilidad en los hombres;
- Menopausia prematura y subfertilidad en las mujeres, aunque existen casos de afectadas que han tenido hijos.
Relación con el Cáncer
Entre las complicaciones de la enfermedad se pueden incluir problemas pulmonares crónicos, diabetes y problemas de aprendizaje sin retraso mental. Sin embargo, la complicación más llamativa de la enfermedad es la susceptibilidad al cáncer. En efecto, la elevada tasa de mutación en el genoma de los afectados con este síndrome conduce a un alto riesgo de cáncer de estas personas. La predisposición al cáncer se caracteriza porque 1) es de amplio espectro, incluyendo leucemias, linfomas y carcinomas, 2) la edad temprana de inicio comparada con el mismo tipo de cáncer en la población general, y 3) la multiplicidad. Las personas con síndrome de Bloom pueden desarrollar cáncer a cualquier edad. La edad media de diagnósticos de cáncer en la cohorte es de aproximadamente 25 años de edad.